1. Read data and get beta values
First, import the necessary packages from pylluminator, and set the logger level to your convenience.
[1]:
from pylluminator.annotations import Annotations, ArrayType, GenomeVersion
from pylluminator.samples import Samples, read_samples
from pylluminator.visualizations import betas_density, betas_dendrogram, betas_2D, nb_probes_per_chr_and_type_hist, plot_mean_beta_diff_per_group
from pylluminator.utils import set_logger, download_from_geo
# path to your data, or a folder where the downloaded data will be saved
data_path = '~/data/pylluminator-tutorial/'
# set the verbosity level, can be DEBUG, INFO, WARNING, ERROR
set_logger('WARNING')
1.1. Read .idat files
1.1.1. Download test data
For the purpose of this tutorial, you can download data from ‘GEO database https://www.ncbi.nlm.nih.gov/geo/’, or skip this step if you want to use you own data.
We downloaded 3 samples from healthy prostate cells (PrEC) and 3 samples from prostate cancer cell (LNCaP)
[2]:
download_from_geo(['GSM7698438', 'GSM7698446', 'GSM7698462', 'GSM7698435', 'GSM7698443', 'GSM7698459'], data_path)
1.1.2. Define and read annotation
First, define the array type of your data and the genome version in order to read the associated information files (manifest, masks, genome information…)
The first run of this code might take a while, as the script will download the manifest and genome info files.
[3]:
# define those parameters to match your data
array_type = ArrayType.HUMAN_EPIC_V2 # run ArrayType? to see available types
genome_version = GenomeVersion.HG38 # run GenomeVersion? to see available genome versions
# read annotation
annos = Annotations(array_type, genome_version, 'updated')
#-------------------------------------------------------------
# alternative: if you don't know what your data is, you can set annos to None and let the script detect the array version when reading samples
# annos = None
1.1.3. Read samples .idat
Now set the paths to your data and to the sample sheet if you have one.
[4]:
# minimum number of detected beads per probe
min_beads = 4
# if no sample sheet is provided, it will be rendered automatically. Use parameter sample_sheet_path or sample_sheet_name to specify one.
my_samples = read_samples(data_path, annotation=annos, min_beads=min_beads, keep_idat=True)
# let's check the generated sample sheet !
my_samples.sample_sheet
[4]:
| sample_id | sample_name | |
|---|---|---|
| 0 | GSM7698443 | PREC_500_2 |
| 1 | GSM7698459 | PREC_500_3 |
| 2 | GSM7698435 | PREC_500_1 |
| 3 | GSM7698446 | LNCAP_500_2 |
| 4 | GSM7698462 | LNCAP_500_3 |
| 5 | GSM7698438 | LNCAP_500_1 |
Now is a good time to check if any sample sheet value has the wrong type. Having a category value encoded as a number can lead to unexpected results, so be sure to convert them to string if need (e.g. the sample ID or sentrix ID)
[5]:
my_samples.sample_sheet.dtypes
# we're all good here, since sentrix_id and sentrix_position are unknown - but if needed here is an example of how to change a column type:
# my_samples = my_samples.astype({'sentrix_id': str})
[5]:
sample_id str
sample_name str
dtype: object
Example of the signal dataframe for a sample
[6]:
my_samples.get_signal_df()['PREC_500_3']
[6]:
| signal_channel | G | R | |||||
|---|---|---|---|---|---|---|---|
| methylation_state | M | U | M | U | |||
| type | channel | probe_type | probe_id | ||||
| I | G | cg | cg00000769_BC11 | 171.0 | 5796.0 | 275.0 | 1561.0 |
| cg00000957_TC11 | 3127.0 | 414.0 | 445.0 | 510.0 | |||
| cg00001349_TC11 | 7365.0 | 2321.0 | 1526.0 | 280.0 | |||
| cg00002033_TC11 | 205.0 | 3947.0 | 195.0 | 378.0 | |||
| cg00002033_TC12 | 232.0 | 4576.0 | 217.0 | 711.0 | |||
| ... | ... | ... | ... | ... | ... | ... | ... |
| II | nan | snp | rs877309_BC21 | 190.0 | NaN | NaN | 8029.0 |
| rs9363764_BC21 | 2458.0 | NaN | NaN | 4125.0 | |||
| rs966367_BC21 | 1264.0 | NaN | NaN | 2420.0 | |||
| rs966367_BC22 | 1117.0 | NaN | NaN | 1991.0 | |||
| rs9839873_BC21 | 879.0 | NaN | NaN | 1363.0 | |||
937688 rows × 4 columns
Let’s add a custom column to our sample sheet
[7]:
my_samples.sample_sheet['sample_type'] = [n.split('_')[0] for n in my_samples.sample_sheet.sample_name]
my_samples.sample_sheet
[7]:
| sample_id | sample_name | sample_type | |
|---|---|---|---|
| 0 | GSM7698443 | PREC_500_2 | PREC |
| 1 | GSM7698459 | PREC_500_3 | PREC |
| 2 | GSM7698435 | PREC_500_1 | PREC |
| 3 | GSM7698446 | LNCAP_500_2 | LNCAP |
| 4 | GSM7698462 | LNCAP_500_3 | LNCAP |
| 5 | GSM7698438 | LNCAP_500_1 | LNCAP |
1.1.4. Save pylluminator samples (optional)
As reading lots of samples can take a couple of minutes, you might want to save the Samples object to a file for later use. Here is how.
[8]:
my_samples.save('raw_samples')
# For later use, here is how to load samples :
# my_samples = Samples.load('raw_samples')
1.2. Calculate and plot beta values
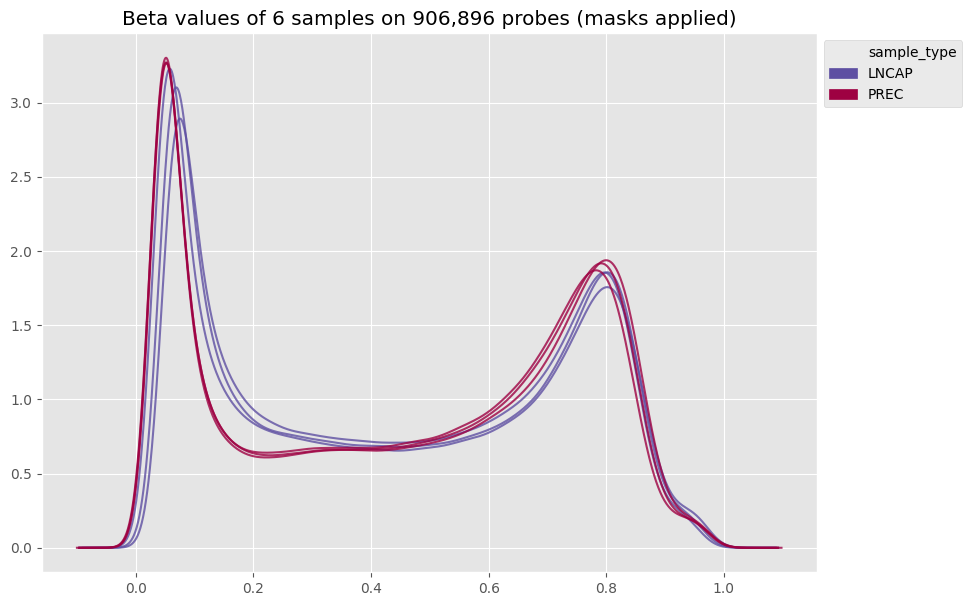
Once your samples are loaded, you can already calculate and plot the beta values of the raw data :
[9]:
my_samples.calculate_betas(include_out_of_band=True)
betas_density(my_samples, color_column='sample_type')
1.3. Preprocessing
Here is the classical preprocessing pipeline for human samples.
Note that each step modifies the Samples object directly, so it’s useful to save the raw samples first (see step 1.4) if you want to try different preprocessing methods.
[10]:
my_samples.mask_quality_probes()
my_samples.infer_type1_channel()
my_samples.dye_bias_correction_nl()
my_samples.poobah()
my_samples.noob_background_correction()
my_samples.calculate_betas(include_out_of_band=True)
[11]:
my_samples.save('preprocessed_samples')
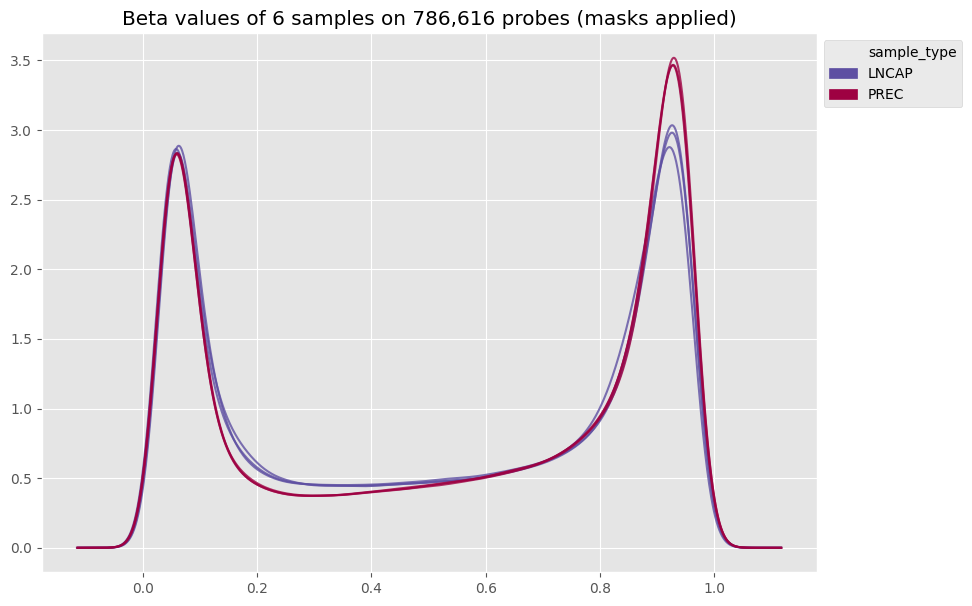
Now let’s see what the preprocessing changed in our beta values!
[12]:
betas_density(my_samples, color_column='sample_type')
1.4. Data insights
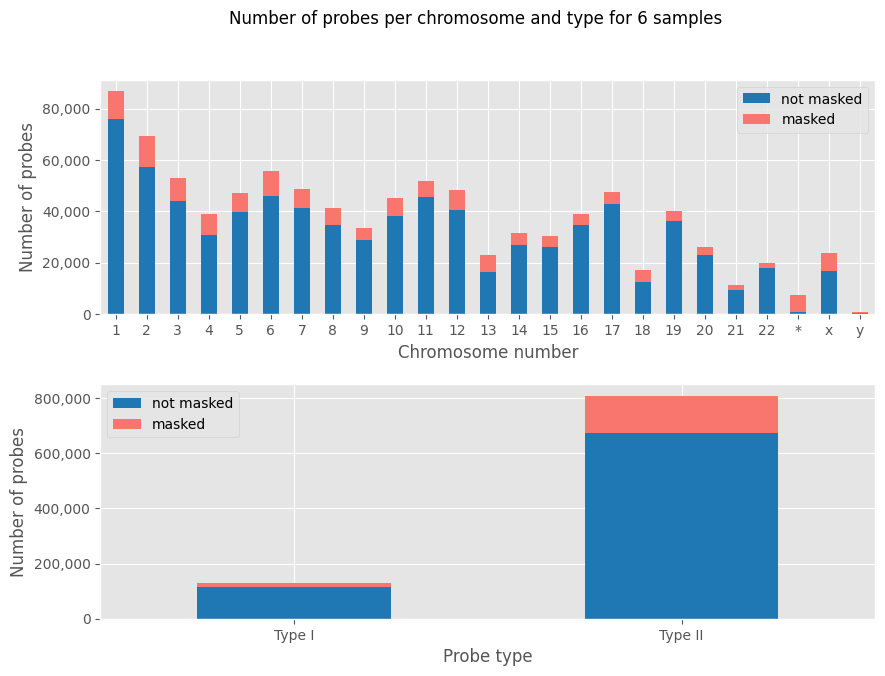
There are a few more plots you can already check to get to know your data:
[13]:
nb_probes_per_chr_and_type_hist(my_samples)
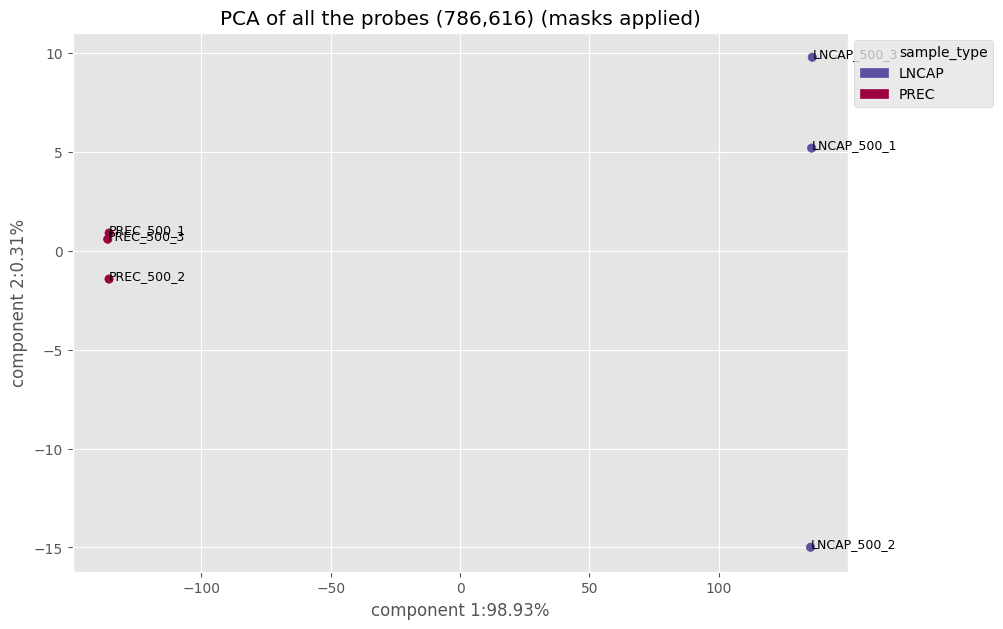
Using a principal component analysis (PCA), we can visualize the spatial distribution of the samples
[14]:
betas_2D(my_samples, model='PCA', color_column='sample_type')
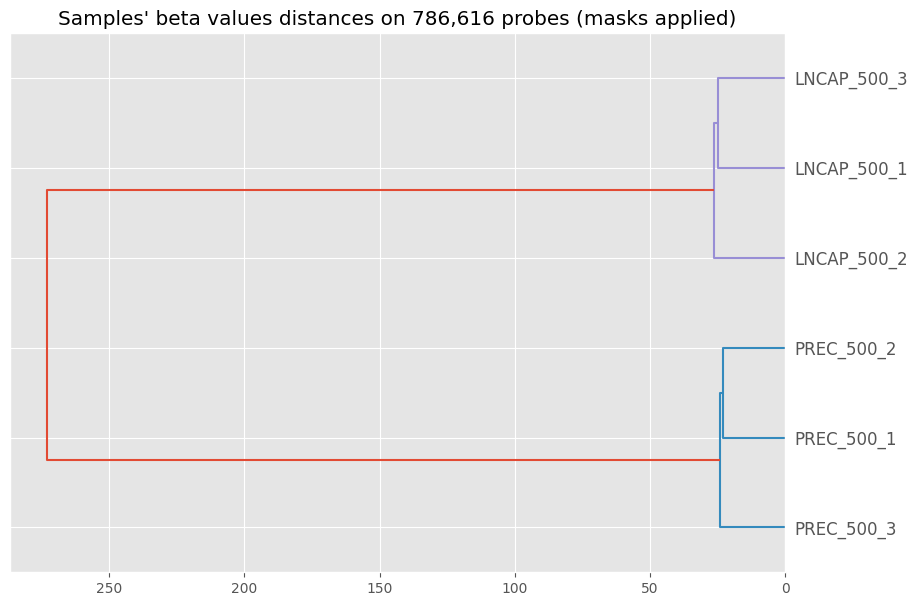
The hierarchical clustering of the samples - nothing really surprising in the output :)
[15]:
betas_dendrogram(my_samples)
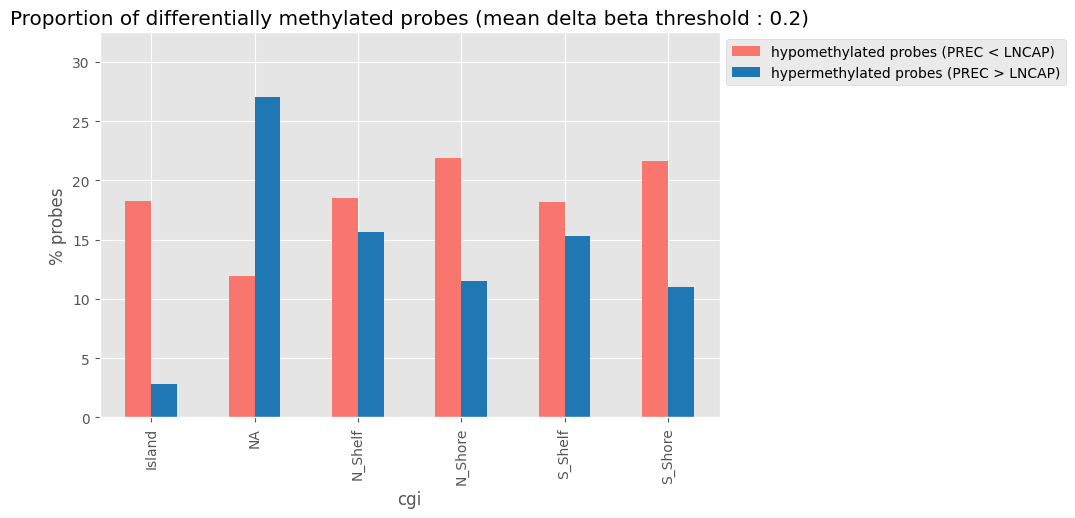
And finally, check the distribution of hypo- and hyper- methylated probes, depending on the probes location
[16]:
plot_mean_beta_diff_per_group(my_samples, group_column='sample_type', figsize=(8, 5))