5. Pathway analysis
Pathway analysis methods provide meaningful insights to better understand how differentially expressed genes (DEG) impact biological processes. Let’s see how we can run Over-Representation Analysis (ORA) and Gene Set Enrichment Analysis (GSEA) on our data, using gseapy package. As the returned objects are pretty big, we delete them at the end of each section for the notebook to save memory.
For more details about the functions used here, you can refer to gseapy documentation, and for more information about GSEA in general (input data etc) : gsea-msigdb website
[1]:
from pylluminator.utils import load_object
from pylluminator.dm import combine_p_values_stouffer
from pylluminator.utils import set_logger
from pylluminator.visualizations import visualize_gene
import numpy as np
import gseapy as gp
import networkx as nx
import matplotlib.pyplot as plt
set_logger('WARNING') # set the verbosity level, can be DEBUG, INFO, WARNING, ERROR
5.1. Data preparation
To run ORA and Pre-rank methods, we need to load DMPs or DMRs. If you haven’t done it yet, check out notebook 3 - DMPs and DMRs.
For GSEA method, we use samples data directly - we use the samples stored in your DM object.
[2]:
my_dms = load_object('dms') # load a DM object
[3]:
# get the genes associated with each probe
annotation_colname = 'genes'
gene_info = my_dms.samples.annotation.probe_infos[['probe_id', annotation_colname]].drop_duplicates().dropna()
# if some probes are associated to several genes, make it one row per gene. Make sure genes are upper case for GSEApy compatibilty
gene_info[annotation_colname] = gene_info[annotation_colname].apply(lambda x: x.upper().split(';'))
gene_info = gene_info.explode(annotation_colname).drop_duplicates()
gene_info.head()
[3]:
| probe_id | genes | |
|---|---|---|
| illumina_id | ||
| 41791408 | cg00000029_TC21 | RBL2 |
| 66725308 | cg00000109_TC21 | FNDC3B |
| 87669537 | cg00000155_BC21 | BRAT1 |
| 47668938 | cg00000158_BC21 | IARS1 |
| 82633489 | cg00000221_BC21 | ANKFN1 |
For each gene, we use the DMPs or the DMRs to compute the fold change (average beta difference between the two conditions) and the significance (by combining the p-values of all associated probes using Stouffer’s method).
[4]:
# chose whether to use DMPs or DMRs
input_type = 'DMP'
# if we work on DMRs, add the probe_ids to the DMRs - for DMPs, just use the DMPs dataframe directly
dm_df = my_dms.dmr.join(my_dms.segments.reset_index().set_index('segment_id')) if input_type == 'DMR' else my_dms.dmp
# add the gene information
dm_df = dm_df.merge(gene_info, on='probe_id')
[5]:
# set the columns to use to select or rank genes (you can check available columns with dm_df.columns)
significance_colname = 'sample_type[T.PREC]_p_value_adjusted'
fold_change_colname = 'avg_beta_delta_sample_type_LNCAP_vs_PREC'
# set the column of the sample sheet that define the type of sample
class_colname = 'sample_type'
# keep only useful columns and remove NAs
dm_df = dm_df[[annotation_colname, fold_change_colname, significance_colname]].dropna()
# aggregate values for each gene
gene_fc_sig_df = dm_df.groupby(annotation_colname).agg({fold_change_colname: 'mean', significance_colname: combine_p_values_stouffer})
gene_fc_sig_df.head()
[5]:
| avg_beta_delta_sample_type_LNCAP_vs_PREC | sample_type[T.PREC]_p_value_adjusted | |
|---|---|---|
| genes | ||
| 5S_RRNA | 0.000252 | 9.888123e-01 |
| 7SK | 0.049935 | 2.797128e-02 |
| A1BG | 0.390756 | 5.746583e-08 |
| A1BG-AS1 | 0.390756 | 5.746583e-08 |
| A2M | 0.004388 | 6.403354e-01 |
For GSEA, we compute the mean beta values per gene, for each sample
[6]:
probes_betas_df = my_dms.samples.get_betas().reset_index().set_index('probe_id').drop(columns=['type', 'channel', 'probe_type'])
genes_betas_df = probes_betas_df.join(gene_info.set_index('probe_id'), how='right').groupby(annotation_colname).mean()
# get the list of sample types, ordered like the beta dataframe
sample_info = my_dms.samples.sample_sheet.set_index(my_dms.samples.sample_label_name)
sample_types = sample_info.loc[probes_betas_df.columns, class_colname]
genes_betas_df.head()
[6]:
| LNCAP_500_1 | LNCAP_500_2 | LNCAP_500_3 | PREC_500_1 | PREC_500_2 | PREC_500_3 | |
|---|---|---|---|---|---|---|
| genes | ||||||
| 5S_RRNA | 0.525830 | 0.505536 | 0.521665 | 0.544796 | 0.539776 | 0.554713 |
| 7SK | 0.412929 | 0.441886 | 0.410811 | 0.159127 | 0.166773 | 0.163154 |
| A1BG | 0.689070 | 0.683857 | 0.660413 | 0.385625 | 0.374149 | 0.381276 |
| A1BG-AS1 | 0.664138 | 0.659627 | 0.637049 | 0.373191 | 0.362245 | 0.369999 |
| A1CF | 0.345105 | 0.355207 | 0.347237 | 0.779690 | 0.784375 | 0.781188 |
[7]:
del dm_df
del probes_betas_df
del gene_info
5.1.1. Gene set selection
You will first need to select the gene set(s) to use in you pathway analysis. You can browse enrichr website or use the gp.get_library_name() function to list available libraries
[8]:
gene_sets = ['GO_Biological_Process_2025', 'KEGG_2021_Human']
5.1.2. Organism specification
Specify the organism you’re working on: Human, Mouse, Yeast, Fly, Fish or Worm
[9]:
organism = 'human'
5.2. Over-Representation Analysis
5.2.1. ORA without background
The minimal input you need to run an ORA is a list of differentially expressed genes (DEG) from your dataset. Here we use the threshold of p-value < 0.05 and abs(fold-change) > 0.2
[10]:
deg = list(set(gene_fc_sig_df[(gene_fc_sig_df[significance_colname] < 0.05) & (abs(gene_fc_sig_df[fold_change_colname]) > 0.2)].index.values))
print(f'Number of genes selected: {len(deg)}/{len(set(gene_fc_sig_df.index))}\n')
enr = gp.enrichr(gene_list=deg, gene_sets=gene_sets, organism=organism)
# output most significant results
enr.results.sort_values('Adjusted P-value').head()
Number of genes selected: 5360/24221
[10]:
| Gene_set | Term | Overlap | P-value | Adjusted P-value | Old P-value | Old Adjusted P-value | Odds Ratio | Combined Score | Genes | |
|---|---|---|---|---|---|---|---|---|---|---|
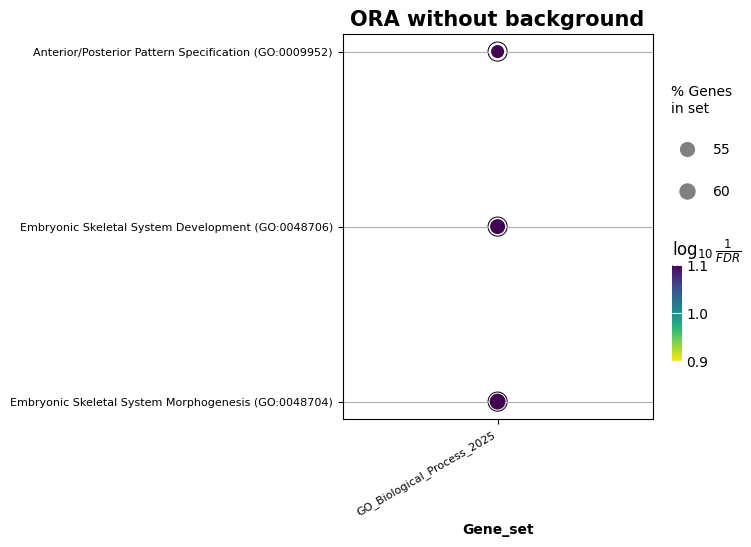
| 0 | GO_Biological_Process_2025 | Embryonic Skeletal System Morphogenesis (GO:00... | 21/33 | 0.000010 | 0.016205 | 0 | 0 | 4.794718 | 55.192524 | TBX1;TFAP2A;OSR2;OSR1;SIX1;TWIST1;SOX11;HOXD11... |
| 1 | GO_Biological_Process_2025 | Embryonic Skeletal System Development (GO:0048... | 24/40 | 0.000010 | 0.016205 | 0 | 0 | 4.110945 | 47.248020 | DLX1;OSR2;DLX3;DLX4;SLC2A10;DLX6;OSR1;WNT5A;SI... |
| 2 | GO_Biological_Process_2025 | Anterior/Posterior Pattern Specification (GO:0... | 34/65 | 0.000011 | 0.016205 | 0 | 0 | 3.008407 | 34.376238 | GATA4;PCSK5;HOXA9;TIFAB;SIX2;HOXA3;HOXC4;HOXA7... |
| 3 | GO_Biological_Process_2025 | Positive Regulation of Morphogenesis of an Epi... | 16/24 | 0.000051 | 0.056812 | 0 | 0 | 5.476048 | 54.128621 | WNT5B;CLSTN1;LIF;SIX1;PAX2;BMP4;GJA1;LBX2;PAX8... |
| 4460 | KEGG_2021_Human | Maturity onset diabetes of the young | 16/26 | 0.000208 | 0.063519 | 0 | 0 | 4.380240 | 37.144421 | PKLR;PDX1;HNF1B;PAX6;BHLHA15;HNF1A;MAFA;GCK;NE... |
[11]:
ax = gp.dotplot(enr.results, x='Gene_set', size=3, top_term=5, title='ORA without background', xticklabels_rot=30, show_ring=True, figsize=(5,5))
# use smaller fonts
for item in ax.get_xticklabels() + ax.get_yticklabels():
item.set_fontsize(8)
ax.xaxis.label.set_size(10)
ax.title.set_size(15)
WARNING:py.warnings:/home/docs/checkouts/readthedocs.org/user_builds/pylluminator/envs/v1.0/lib/python3.12/site-packages/gseapy/plot.py:753: FutureWarning: DataFrameGroupBy.apply operated on the grouping columns. This behavior is deprecated, and in a future version of pandas the grouping columns will be excluded from the operation. Either pass `include_groups=False` to exclude the groupings or explicitly select the grouping columns after groupby to silence this warning.
.apply(lambda _x: _x.sort_values(by=self.colname).tail(self.n_terms))
5.2.2. ORA with background
By default, selected genes are tested against all genes available in the gene sets. For better results, it can be relevant to narrow it down to a list of genes of interest.
Let’s use the genes that were detected in our experiment as background genes, and see how it changes the result.
[12]:
background_genes = list(set(gene_fc_sig_df.index))
print('Number of selected genes: ', len(deg))
print('Number of background genes: ', len(background_genes), '\n')
enr_bg = gp.enrichr(gene_list=deg, gene_sets=gene_sets, organism=organism, background=background_genes)
# output top 5 results results
enr_bg.results.sort_values('Adjusted P-value')[:5]
Number of selected genes: 5360
Number of background genes: 24221
[12]:
| Gene_set | Term | P-value | Adjusted P-value | Old P-value | Old adjusted P-value | Odds Ratio | Combined Score | Genes | |
|---|---|---|---|---|---|---|---|---|---|
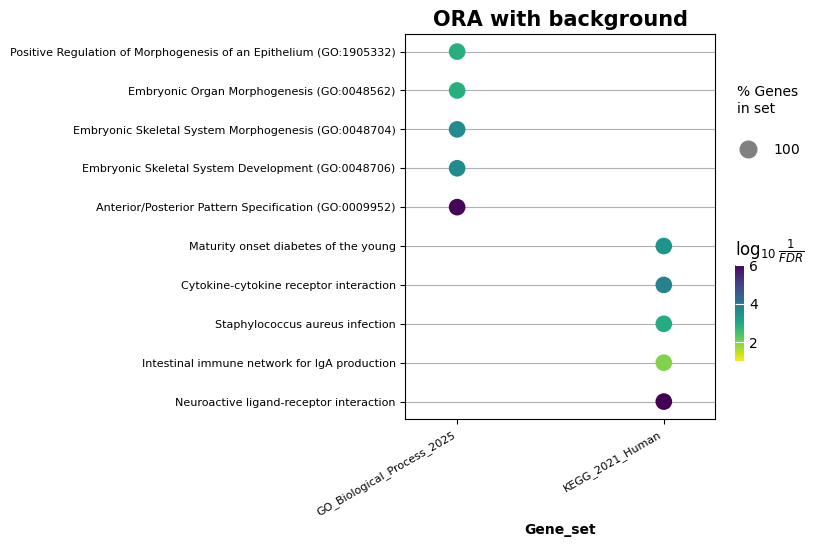
| 4460 | KEGG_2021_Human | Neuroactive ligand-receptor interaction | 2.909977e-09 | 8.904529e-07 | 0 | 0 | 2.199438 | 43.230221 | OXTR;NPFFR1;CHRM1;SCT;RXFP4;NR3C1;RXFP1;RXFP3;... |
| 0 | GO_Biological_Process_2025 | Anterior/Posterior Pattern Specification (GO:0... | 2.683214e-10 | 1.196713e-06 | 0 | 0 | 5.727161 | 126.219950 | GATA4;PCSK5;HOXA9;TIFAB;SIX2;HOXA3;HOXC4;HOXA7... |
| 4461 | KEGG_2021_Human | Cytokine-cytokine receptor interaction | 1.093437e-06 | 1.672958e-04 | 0 | 0 | 2.213999 | 30.389760 | ACVRL1;CXCL6;CD40;IL25;TNFRSF13B;CSF1;EPO;IL27... |
| 1 | GO_Biological_Process_2025 | Embryonic Skeletal System Development (GO:0048... | 1.311706e-07 | 2.442377e-04 | 0 | 0 | 5.650975 | 89.549675 | DLX1;OSR2;DLX3;DLX4;SLC2A10;DLX6;OSR1;WNT5A;SI... |
| 2 | GO_Biological_Process_2025 | Embryonic Skeletal System Morphogenesis (GO:00... | 1.642854e-07 | 2.442377e-04 | 0 | 0 | 6.740282 | 105.294391 | TBX1;TFAP2A;OSR2;OSR1;SIX1;TWIST1;SOX11;HOXD11... |
Visualize the top 5 terms for each gene set, with the default significance cutoff set at 0.05
[13]:
ax = gp.dotplot(enr_bg.results, x='Gene_set', size=2, top_term=5, figsize=(5, 5), y_order=True, title = 'ORA with background', xticklabels_rot=30)
# use smaller fonts
for item in ax.get_xticklabels() + ax.get_yticklabels():
item.set_fontsize(8)
ax.xaxis.label.set_size(10)
ax.title.set_size(15)
WARNING:py.warnings:/home/docs/checkouts/readthedocs.org/user_builds/pylluminator/envs/v1.0/lib/python3.12/site-packages/gseapy/plot.py:753: FutureWarning: DataFrameGroupBy.apply operated on the grouping columns. This behavior is deprecated, and in a future version of pandas the grouping columns will be excluded from the operation. Either pass `include_groups=False` to exclude the groupings or explicitly select the grouping columns after groupby to silence this warning.
.apply(lambda _x: _x.sort_values(by=self.colname).tail(self.n_terms))
[14]:
del deg
del enr
del enr_bg
5.3. Pre-rank GSEA
As we have already calculate DMPs an DMRs, we can use them to rank the genes and use the pre-rank method provided by gseapy.
Here we use the formula sign(FC) * - log10(significance) to rank the genes, that bring the most upregulated genes at the beginning of the list, the most downregulated at the end, and the less differentialy expressed genes in the middle.
[15]:
def rank_formula(row):
if row[significance_colname] == 0:
return np.sign(row[fold_change_colname]) * np.inf
return np.sign(row[fold_change_colname]) * -np.log10(row[significance_colname])
rank_data = gene_fc_sig_df.apply(rank_formula, axis=1).sort_values(ascending=False)
[16]:
# we chose a low permutation number to speed up the demo
pre_res = gp.prerank(rnk=rank_data, gene_sets=gene_sets, verbose=True, permutation_num=1000, max_size=300, threads=4)
2025-09-16 14:15:56,017 [WARNING] Input gene rankings contains inf values!
WARNING:py.warnings:/home/docs/checkouts/readthedocs.org/user_builds/pylluminator/envs/v1.0/lib/python3.12/site-packages/gseapy/gsea.py:507: FutureWarning: The 'method' keyword in Series.replace is deprecated and will be removed in a future version.
rankser.replace(-np.inf, method="ffill", inplace=True)
WARNING:py.warnings:/home/docs/checkouts/readthedocs.org/user_builds/pylluminator/envs/v1.0/lib/python3.12/site-packages/gseapy/gsea.py:508: FutureWarning: The 'method' keyword in Series.replace is deprecated and will be removed in a future version.
rankser.replace(np.inf, method="bfill", inplace=True)
2025-09-16 14:15:56,020 [WARNING] Duplicated values found in preranked stats: 13.22% of genes
The order of those genes will be arbitrary, which may produce unexpected results.
2025-09-16 14:15:56,021 [INFO] Parsing data files for GSEA.............................
2025-09-16 14:15:56,154 [INFO] Downloading and generating Enrichr library gene sets......
2025-09-16 14:16:04,451 [INFO] Downloading and generating Enrichr library gene sets......
2025-09-16 14:16:05,822 [INFO] 3090 gene_sets have been filtered out when max_size=300 and min_size=15
2025-09-16 14:16:05,824 [INFO] 2573 gene_sets used for further statistical testing.....
2025-09-16 14:16:05,825 [INFO] Start to run GSEA...Might take a while..................
2025-09-16 14:19:09,738 [INFO] Congratulations. GSEApy runs successfully................
Order the pathways by their False Discovery Rate (FDR), and show the ones that have a FDR lower than 0.05
[17]:
pre_res.res2d = pre_res.res2d.sort_values('FDR q-val').reset_index(drop=True)
pre_res.res2d[pre_res.res2d['FDR q-val'] < 0.05]
[17]:
| Name | Term | ES | NES | NOM p-val | FDR q-val | FWER p-val | Tag % | Gene % | Lead_genes | |
|---|---|---|---|---|---|---|---|---|---|---|
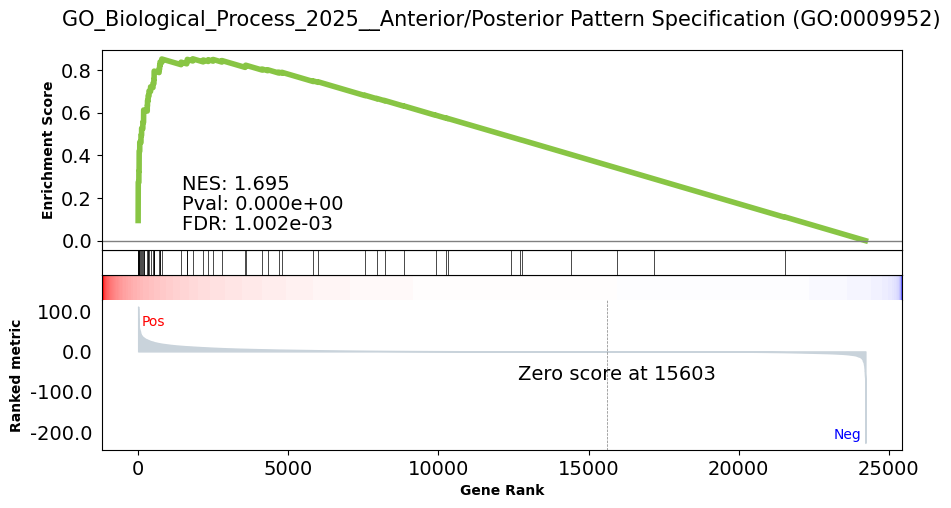
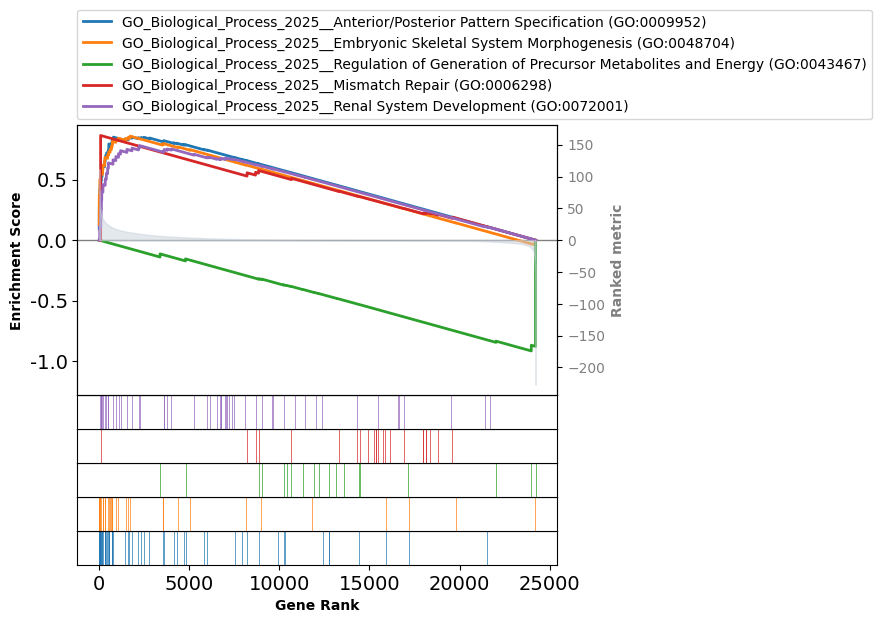
| 0 | prerank | GO_Biological_Process_2025__Anterior/Posterior... | 0.852806 | 1.694523 | 0.0 | 0.001002 | 0.001 | 29/55 | 7.51% | HOXD3;HOXA3;HOXB3;WT1;HOXC4;HOXA9;HOXC6;GATA4;... |
| 1 | prerank | GO_Biological_Process_2025__Embryonic Skeletal... | 0.860125 | 1.671863 | 0.0 | 0.003006 | 0.006 | 21/32 | 7.09% | HOXD3;HOXA3;HOXB3;HOXC4;HOXA9;COL2A1;HOXC9;SIX... |
| 2 | prerank | GO_Biological_Process_2025__Regulation of Gene... | -0.915732 | -2.12952 | 0.0 | 0.025516 | 0.025 | 2/19 | 0.99% | PRDM16;PRKAG2 |
| 3 | prerank | GO_Biological_Process_2025__Mismatch Repair (G... | 0.865357 | 1.609492 | 0.0 | 0.042411 | 0.112 | 1/24 | 0.33% | TP73 |
Now we can visualize the most significant term
[18]:
term = pre_res.res2d.Term[0]
fig = pre_res.plot(terms=term, figsize=(10, 5))
# use smaller fonts
for ax in fig.axes:
ax.set_xlabel(ax.get_xlabel(), fontsize=10)
ax.set_ylabel(ax.get_ylabel(), fontsize=10)
_ = fig.suptitle(term, fontsize=15)
Or visualize the top 5 terms on the same plot:
[19]:
fig = pre_res.plot(terms=pre_res.res2d.Term[:5],figsize=(3,4))
# use smaller fonts
for ax in fig.axes:
ax.set_xlabel(ax.get_xlabel(), fontsize=10)
ax.set_ylabel(ax.get_ylabel(), fontsize=10)
[20]:
ax = gp.dotplot(pre_res.res2d, column='FDR q-val', title='Pre-ranked GSEA', cmap=plt.cm.viridis, size=3, figsize=(3, 4), show_ring=True)
# use smaller fonts
for item in ax.get_xticklabels() + ax.get_yticklabels():
item.set_fontsize(8)
ax.xaxis.label.set_size(10)
ax.title.set_size(15)

[21]:
term_idx = 0 # chose the first term
genes = pre_res.res2d.Lead_genes[term_idx].split(";")
ax = gp.heatmap(df = genes_betas_df.loc[genes], z_score=0, title=pre_res.res2d.Term[term_idx], figsize=(7,6), yticklabels=False)
# use smaller fonts and show all labels
genes.reverse() #for labels to be in the right order
ax.title.set_size(15)
_ = ax.set_xticks(range(len(genes_betas_df.columns)), labels=genes_betas_df.columns, rotation=30, ha="center", fontsize=8)
_ = ax.set_yticks(range(len(genes)), labels=genes, va="bottom", fontsize=8)
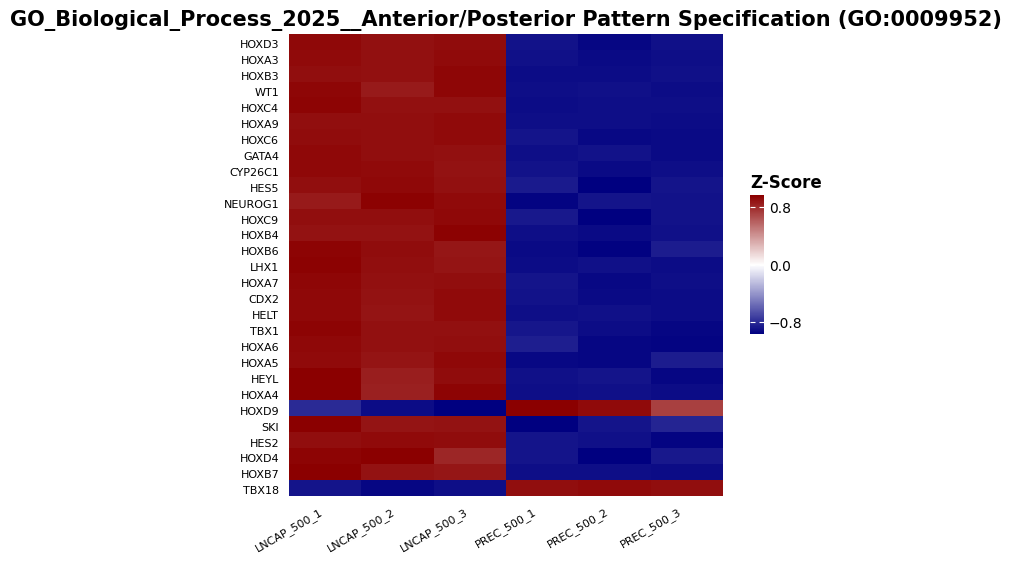
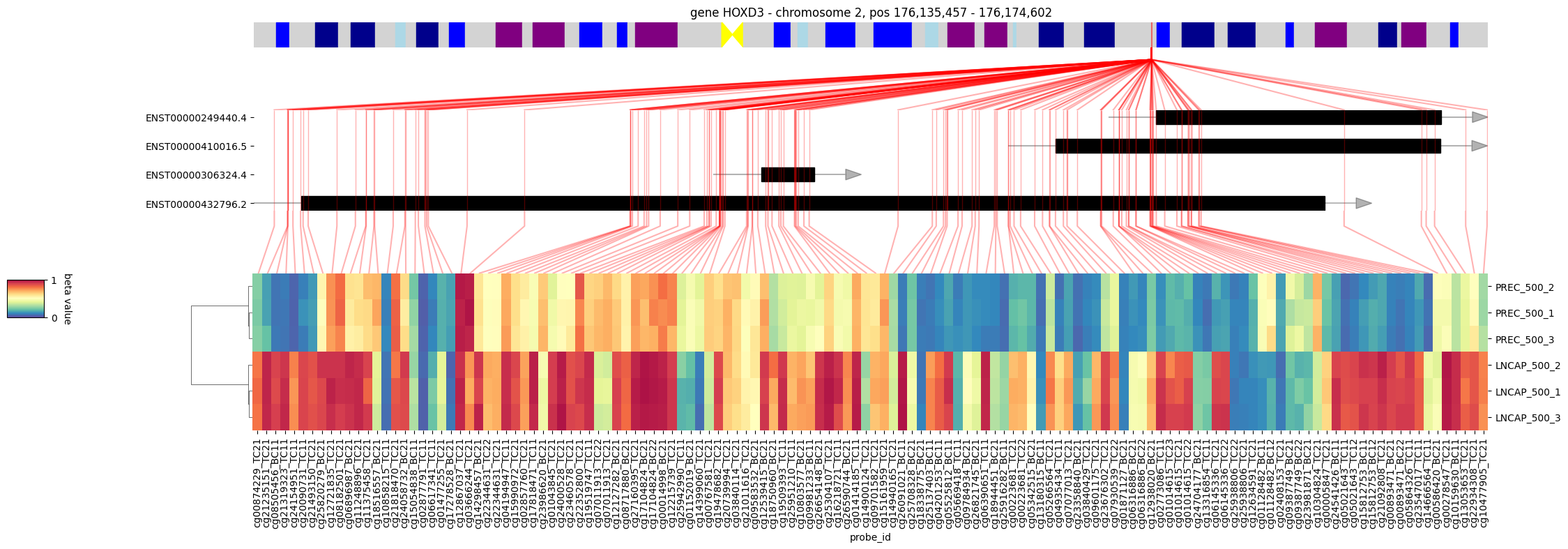
Using pylluminator’s function, we can go back to checking the beta values of the probes associated to a given gene
[22]:
visualize_gene(my_dms.samples, 'HOXD3', figsize=(20, 6))
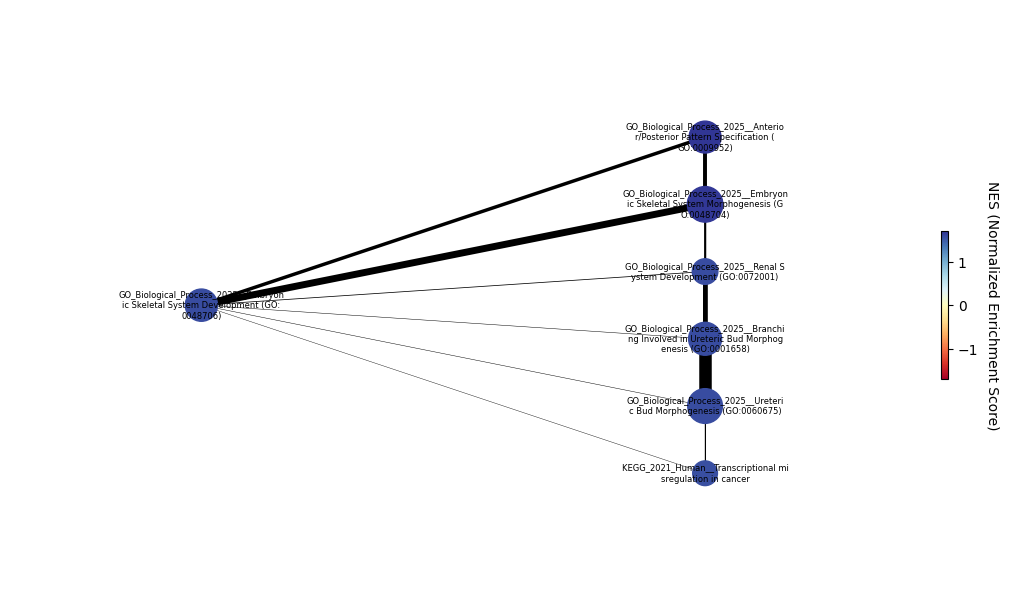
Finally, compute the enrichment map to show the relations between the detected relevant pathways
[23]:
nodes, edges = gp.enrichment_map(pre_res.res2d, cutoff=0.1) # chose a higher p-value cutoff to show more nodes, default is 0.05
# build graph
G = nx.from_pandas_edgelist(edges,
source='src_idx',
target='targ_idx',
edge_attr=['jaccard_coef', 'overlap_coef', 'overlap_genes'])
nodes = nodes.loc[G.nodes.keys()] # remove nodes that are not connected by any edge
fig, ax = plt.subplots(figsize=(10, 6), layout='constrained')
# init node coordinates
pos=nx.layout.bfs_layout(G, start=nodes.index[0])
# add a colorbar
nodes_cmap = plt.cm.RdYlBu
max_NES = max(abs(min(nodes.NES)), max(nodes.NES))
sm = plt.cm.ScalarMappable(cmap=nodes_cmap, norm=plt.Normalize(vmin=-max_NES, vmax=max_NES))
sm.set_array([])
cbar = plt.colorbar(sm, ax=ax, shrink=0.25)
cbar.set_label('NES (Normalized Enrichment Score)', rotation=270, labelpad=15)
# draw nodes
nx.draw_networkx_nodes(G, pos=pos,
cmap=nodes_cmap, node_color=nodes.NES, vmin=-max_NES, vmax=max_NES,
margins=0.3, node_size=list(nodes.Hits_ratio *1000))
# draw node labels - wrap labels so that they don't overlap
max_length = 35
wrapped_labels = {k: "\n".join([v[i:i+max_length] for i in range(0, len(v), max_length)]) for k, v in nodes.Term.to_dict().items()}
nx.draw_networkx_labels(G, pos=pos, font_size= 6, labels=wrapped_labels)
# draw edges
edge_weight = nx.get_edge_attributes(G, 'jaccard_coef').values()
nx.draw_networkx_edges(G, pos=pos, width=list(map(lambda x: x*10, edge_weight)))
plt.axis('off')
plt.show()
[24]:
del gene_fc_sig_df
del pre_res
del my_dms
5.4. GSEA
Since we are working with two groups (healthy control cells and prostate cancer cells), we can directly use the GSEA function on our ‘raw’ data (here, the genes beta values) with the corresponding phenotype dataframe (sample_types), to find the enriched pathways. Uncomment the following code to run the analysis.
[25]:
# gs = gp.GSEA(data=genes_betas_df.dropna(), gene_sets=gene_sets, classes=sample_types, permutation_num=1000)
# gs.pheno_neg = 'PREC' # control samples
# gs.pheno_pos = 'LNCAP' # samples of prostate cancer cells
# gs.run()
Visualize the top 5 identified pathways
[26]:
# _ = gs.plot(gs.res2d.Term[:5])
Visualize, for a given pathway and for each gene, the samples z-score.
[27]:
# term_idx = 0 # chose the first term
# genes = gs.res2d.Lead_genes[term_idx].split(";")
# ax = gp.heatmap(df = genes_betas_df.loc[genes], z_score=0, title=gs.res2d.Term[term_idx], figsize=(8,4))